
As word spread from India last year that common bacteria strains showed resistance to all known antibiotics, some officials feared that terrorists might find a way to weaponize those bacteria and trigger an epidemic. A later study revealed these bacteria were in virtually every modern country’s drinking water supply. Officials worried that if people picked up one of these bugs, became ill and tried taking antibiotics to recover, the bacteria could bypass any drug.
To produce the first publicly available model of a potent enzyme that was making the bacteria into superbugs, groups from across the Department of Energy’s Argonne National Laboratory and collaborators at Texas A&M University plied the computational muscle of Intrepid, the lab’s IBM Blue Gene/P.
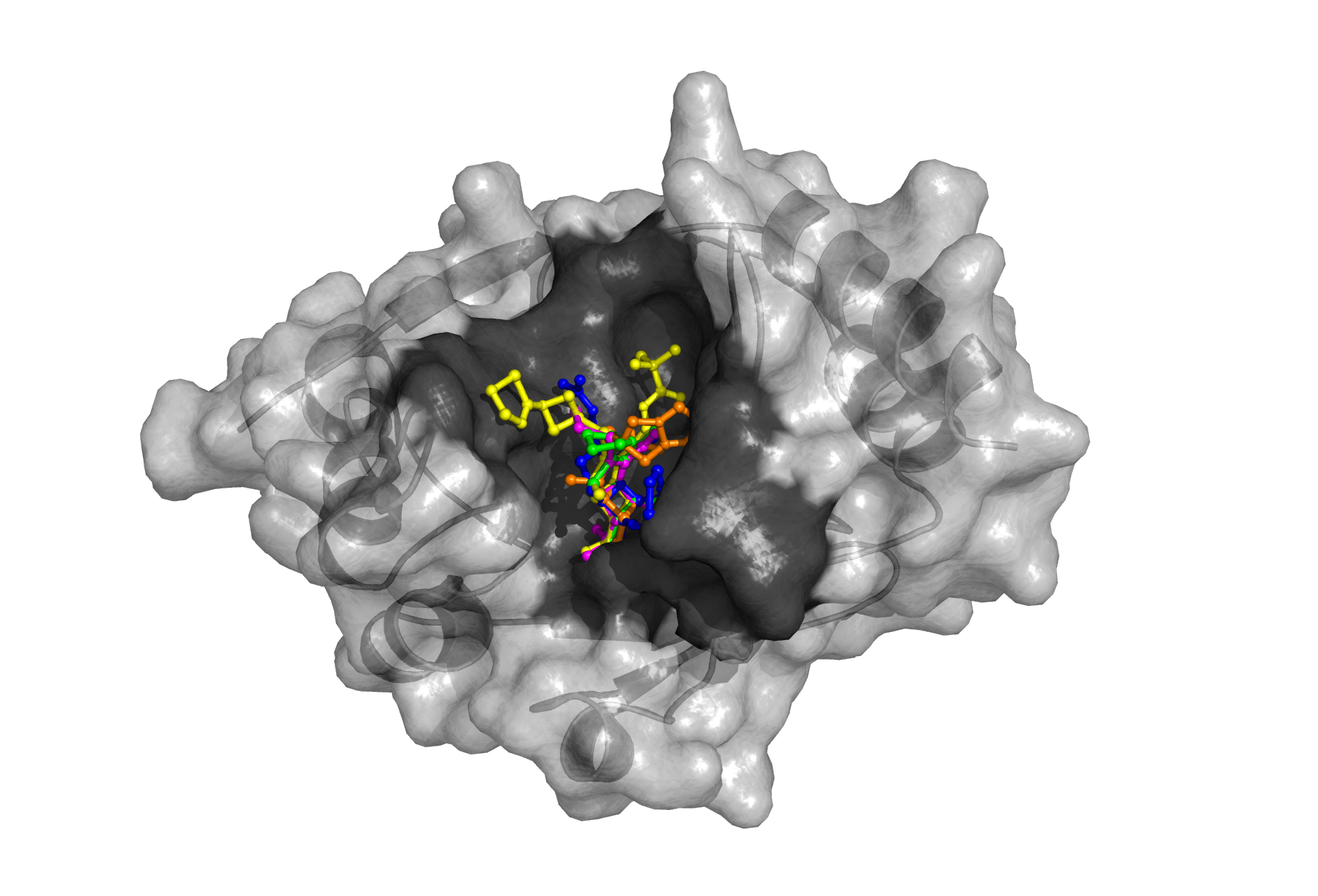
Researchers focused on the bacterial enzyme NDM-1 (New Delhi Metallo-β-lactamase), which grabs a piece of the antibiotic beta-lactam ring, breaks it and renders the antibiotic useless. Analysis revealed the enzyme has an enormous and flexible active site, capable of gobbling the stout rings of existing antibiotics, including those of many powerful carbapenems, antibiotics of last resort. Decoding NDM-1 and quickly publishing their findings, the Argonne team documented the enzymes’ promiscuity and uncovered strategic flaws in existing antibiotic development strategies.
“From our simulations it appears that NDM-1 is capable of processing much larger molecules, voiding the easy path” of modifying existing molecules “to breathe new life into old drugs,” says Andrew Binkowski, assistant scientist in Argonne’s biosciences division and a fellow at the University of Chicago’s Computation Institute.
“The NDM-1 story is a really good test case of speed to solution,” says Binkowski, who collaborates with a team from Argonne, the University of Chicago and Northwestern University. “The time to the final solution is really important. If there is an outbreak and we want to quickly find viable drug leads to treat the infection, we can’t afford to wait hundreds of days. We want it in a matter of hours. Intrepid enables that possibility. We went in short order from headlines about a new national security threat to a crystallographic model of the protein used for designing new antibiotics, just months later.”
Binkowski’s computational models analyze the three-dimensional structure of important classes of protein targets – stretches of DNA on a protein that look promising as docking sites for antibiotics and drugs. The National Institutes of Health, DOE and researchers from across the scientific community suggest protein-target nominees. Binkowski also is a member of the Center for Structural Genomics in Infectious Diseases (CSGID), which is supported by the NIH’s National Institute of Allergy and Infectious Diseases. The center applies technologies for rapidly analyzing biological structures to improve drugs attacking organisms that cause infectious diseases. CSGID and Argonne’s Midwest Center for Structural Genomics, which took part in the NDM-1 study, are considered among the world’s most productive hubs of their kind, deciphering and analyzing hundreds of protein structures each year.
Ligands in search of proteins
Binkowski concentrates on protein targets and ligand relationships. A protein is a macromolecule that might have tens of thousands of atoms; the ligand is a smaller molecule with fewer than 100 atoms. Think of the big protein sitting on a sort of dock. The ligand is a boat seeking a favorable place to land and unload goods, such as antibiotics.
When the ligand gets close enough, forces from the protein’s amino acid residues begin to act on it, Binkowski explains. “In normal protein activity this allows enzymes to perform their normal functions, catalyzing chemical reactions. In infectious diseases, we want to prevent or alter the organism’s normal functions, so we want to design a small molecule that will bind more aggressively to the protein. The more we know about the protein structure, the more effective we can be at doing this.”
Running on Intrepid, Binkowski uses an automated pipeline to model many different ligands and their interactions with a protein. It starts with crude docking models and, employing more sophisticated software that asks more stringent questions at each step, concludes with a list of candidate ligands for protein target sites.
“I have a whole suite of software applications that enables me to simulate and analyze the 3-D structure of the protein and to identify possible sites that could be important or capable of binding different small molecules,” he says. “I also speculate what kinds of molecules might be attracted to this particular spot. Moreover, if we find some molecules that should be attracted to the target, we ask, ‘What kinds of modifications can we do to make them even more attracted and better binders?’”
Supercomputer modeling establishes the ground floor for rational drug design – that is, working from a hypothesis based on a drug target’s biology rather than trial and error. Later, when he thinks he has the right match of protein target and ligand, Binkowski and his associates test their computational predictions by measuring binding in living organisms. They also use X-ray crystallography experiments, with the aid of Argonne’s Structural Biology Center at the powerful Advanced Photon Source, to validate their biomolecular simulations.