
Electric cars will remain a tough sell until they can travel beyond 100 miles before needing to recharge their batteries. The battery-life bottleneck has driven the search for technologies that extend the energy storage capacity of lithium-ion batteries.
A collaboration, led by Gao Liu of Lawrence Berkeley National Laboratory’s Environmental Energy Technologies Division and Wanli Yang of Berkeley’s Advanced Light Source, has developed a next-generation battery that could fill the need.
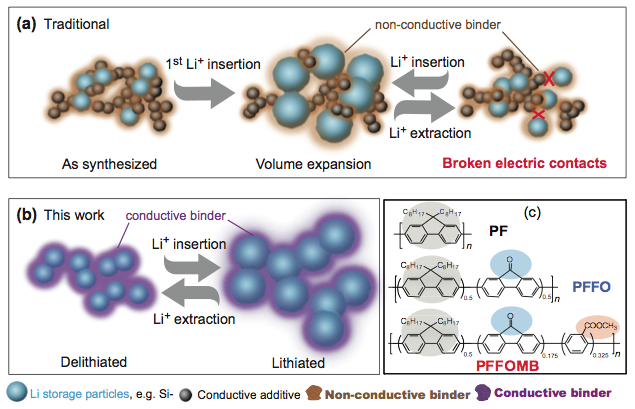
Combining computational modeling and advanced materials synthesis, the Berkeley Lab scientists sought a low-cost anode to provide the needed battery boost. Their solution involves replacing inert graphite with silicon nanoparticles bound to a polymer that absorbs eight times the lithium and becomes electrically conductive as it does. The researchers hope the advance will help power the next generation of electronics and extend the range of electric vehicles.
They key to the advance was making silicon a practical anode material.
Scientists have known for decades that a silicon atom can absorb almost four lithium atoms. But in doing so, it balloons to three times its size. The shrinking and swelling during each round of discharging and charging have made silicon an impractical choice for battery anodes.
Materials scientists have tried a work-around by forming the silicon into nanoparticles and connecting them with an inert polymer binder and a graphite electrical conductor to improve performance. After a few charging and discharging cycles, however, the graphite tends to lose contact with the silicon nanoparticles, reducing its conductivity.
The new-generation polymer acts as both a mechanical binder and an electrical conductor. After testing several variations, the Berkeley Lab group designed a variant of a polyfluorine-based polymer hat worked much better than a previously tested conducting polymer. They hypothesized that a particular modification to the variant polymer – the addition of a carbonyl group (a double-bonded carbon-oxygen group) – contributed to its unique electronic properties. Indeed, the researchers used Berkley’s Advanced Light Source to produce an X-ray absorption spectrum that showed there is an additional peak below the electron conduction band. But that didn’t explain how the carbonyl group and an associated additional X-ray absorption peak were related to the enhanced battery performance.
To solve this mystery, they turned to Lin-Wang Wang, a computational scientist in Berkeley’s Materials Science Division. Wang has spent many years developing and testing algorithms that employ density functional theory (DFT), a method that simulates the electronic properties of systems starting from fundamental physics. The battery system perfectly fit those that DFT simulates well, Wang says. It was an ordered system with structures built on the nanoscale, including the polymer, silicon nanoparticles and lithium ions.
According to Wang, several possible scenarios could account for the increased conductance and the X-ray absorption findings. By using codes that implement the density functional theory, and the computational resources at the Berkeley Lab-based National Energy Research Science Computing Center (NERSC), Wang and postdoctoral fellow Nenad Vukmirovic modeled lithium interaction with the silicon-polymer electrode.
“Of the many possible explanations of the experimental results, density functional theory pointed out how the whole thing works,” Wang says.
The theoretical calculations dovetailed nicely with the experimental calculations, providing a detailed look at how and why the polymer works so well.
Lithium ions – rather than handing off electrons to the embedded nanoparticle silicon – donate electrons to the polymer first, providing it with its conducting properties. Only afterward do lithium ions adhere to the silicon. Because of its conductivity, the polymer acts as an electrical bridge, allowing the positively charged lithium ions to move into the silicon and combine with the negatively charged electrons flowing through the polymer, something that no other previously tested polymer could do. The findings, published in the September 2011 issue of the journal Advanced Materials, provided an experimental and theoretical foundation for further refinement and advancement of battery components.
The ability to incorporate DFT calculations into the new materials development process was a relatively new phenomenon, Wang says. Ten years ago, DFT algorithms were untested and computationally intensive. It took many years to demonstrate that results obtained by DFT were reliable. But in recent years, the method has proven to be a valuable tool to provide insight into the underlying physics, particularly of nanostructural materials, Wang says.
The next generation of DFT algorithms, which can provide more accurate results and can calculate bigger systems, combined with the computational resources at NERSC and other facilities, will allow DFT to be almost a routine tool to explain and predict properties of new nanomaterials. Wang expects DFT to contribute to the development of new quantum dots and organic polymer arrays by tracking individual electrons as they move through the system.
In the case of the battery simulation, DFT provided a complete picture of the lithium-polymer interaction.
“At the beginning of this project, we discussed many wild pictures that were all possible,” Wang says. “It is really only through this calculation that we pinned down the actual mechanisms of how the system works.”
The picture that finally emerged, Wang adds, was “very beautiful.”